Это наследственная патология, предрасполагающая к возникновению опухолей у человека. Выделяют семь типов нейрофиброматоза, среди которых наибольшую значимость имеют заболевание I типа (болезнь Реклингхаузена) и II типа (с двусторонними невриномами 8 пары черепных нервов). Патология сопровождается формированием опухолей в области нервных тканей, что провоцирует появление костных и кожных аномалий.
В половине случаев болезнь является наследственной и относится к аутосомно-доминантной группе. В 50 % случаев ее связывают со спонтанной мутацией, происходящей в гене НФ-1 на длинном плече хромосомы 17-й пары. При нейрофиброматозе в этой области присутствуют различные типы мутаций и перестроек, в т.ч. делеции, инверсии, транслокации, точковые мутации. В 80 % случаев они ведут к синтезу дефектного белка нейрофибромина, что становится причиной возникновения новообразований.
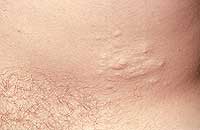
Ранний признак заболевания — множественные мелкие овальные пигментные пятна с желтовато-коричневым цветом (оттенок кофе с молоком) и гладкой поверхностью. При нейрофиброматозе они локализуются преимущественно в области подмышек, паховых складок.
Также болезнь проявляет себя узелками Лиша, которые представляют собой гамартомы (белесоватые пятна) на радужной оболочке глаза, которые не видны невооруженным взглядом.
Характерный признак нейрофиброматоза — наличие кожных и плексиформных нейрофибром. Кожные образования представляют собой ограниченные доброкачественные опухоли, которые располагаются подкожно и образуются на оболочках мелких нервов кожного покрова. Плексиформные нейрофибромы, как правило, имеют больший размер, возникают на крупных нервах и вызывают нарушение их функций.
Нейрофиброматоз сопровождается развитием опухолей ЦНС, чаще всего глиом зрительных нервов, астроцитом, эпендимом, неврином слухового нерва, менингиом и нейрофибром.
При выраженной форме патологии могут развиваться сколиоз, дефекты тел позвонков и их отростков, увеличение межпозвоночных отверстий, эрозии их краев, а также краевые дефекты задних отделов ребер. Длинные трубчатые кости скелета при нейрофиброматозе атрофируются, изгибаются, либо гипертрофируются и утолщаются. Возможна асимметрия костей черепа.
Среди прочих симптомов патологии: когнитивные нарушения, эпилептические припадки, нарушения поведения, эндокринные расстройства, снижение мышечного тонуса, сирингомиелия, стеноз почечной и легочной артерии и др.
Диагноз подтверждается при наличии у пациента двух и более из следующих признаков:
- шесть и более характерных пигментных пятен (диаметр 5 мм у детей и 15 мм у взрослых);
- две и более кожных нейрофибромы и одна плексиформная нейрофиброма;
- два и более узелка Лиша;
- гиперпигментация кожи в области подмышек и паха;
- костные аномалии;
- глиомы зрительных нервов;
- нейрофиброматоз у ближайших родственников.
Генетическое тестирование позволяет узнать о наличии аномалий в геноме, способных привести к данному заболеванию. В медико-генетическом центре «Геномед» можно пройти такое обследование.
Для лечения нейрофиброматоза используются препараты, способствующие нормализации определенных обменных процессов в организме. Косметические дефекты устраняются хирургическим путем.
Среди показаний к операции выделяют:
- резкую болезненность, изъязвление опухоли;
- затрудненность движений из-за нейрофибром;
- сдавление или смещение внутренних органов вследствие присутствия опухоли.
источник
Самое распространенное наследственное заболевание, предрасполагающее к возникновению опухолей у человека. Является аутосомно-доминантным, встречается с одинаковой частотой у мужчин и женщин у 1 из примерно 3500 новорожденных. В 50% случаев заболевание является наследственным, в 50% – результатом спонтанной мутации.
НФ1 обладает полной (100%) пенетрантностью, т.е. больны все носители патологического гена, но экспрессия гена, т.е. степень выраженности вызванных генным дефектом нарушений, очень вариабельна, даже в одной семье могут наблюдаться как минимально выраженные, так и тяжелые случаи. Риск наследования ребенком патологического гена составляет 50% при наличии НФ1 у одного из родителей и 66,7% — у обоих.
Во всех случаях НФ1 генетический дефект локализуется в зоне 11.2 хромосомы 17 (17q11.2).Расположенный здесь ген НФ1 кодирует синтез крупного белка — нейрофибромина.
При повреждении гена НФ1 в одной из хромосом 17 пары 50% синтезируемого нейрофибромина становятся дефектными, и наблюдается смещение равновесия роста клеток в сторону пролиферации.
Диагноз НФ1 устанавливается на основании выявления двух и более из ниже перечисленных признаков:
— 6 и более пятен цвета “кофе с молоком” на коже, более 5 мм в диаметре у ребенка или 15 мм у взрослого, видимых при обычном комнатном освещении.
— 2 и более нейрофибром любого типа.
— Гиперпигментации подмышек или паховой области.
— Глиомы зрительных нервов.
— 2 и более узелков Лиша (пигментированных гамартом радужки).
— Костных аномалий (истончения кортикального слоя трубчатых костей, часто приводящего к формированию ложных суставов).
— Наличия прямого родственника с НФ1.
Следует подчеркнуть, что указанные признаки могут встречаться в любом сочетании, но ни один из них сам по себе не является достаточным для диагностики НФ1. С учетом сказанного, даже отсутствие пятен цвета «кофе с молоком» (выявляемых у 99% больных НФ1) не противоречит диагнозу НФ1 при наличии двух или более других проявлений этого заболевания.
При НФ1 за счет нарушения регуляции клеточного роста возникает ряд ассоциированных состояний (или заболеваний). К ним относятся:
— Шванномы любого нерва (но не двусторонние вестибулокохлеарного).
— Спинальные и(или) периферические нейрофибромы.
— Множественные кожные нейрофибромы.
— Макроцефалия.
— Опухоль головного мозга.
— Односторонний дефект крыши орбиты – пульсирующий экзофтальм.
— Кифосколиоз.
— Висцеральные проявления вследствие вовлечения нервов.
— Сирингомиелия.
— Злокачественные опухоли (чаще встречаются MPNST, ганглиоглиома, саркома, лейкемия, нефробластома (опухоль Вильма)).
— Феохромоцитомы.
— Соматические заболевания или состояния, прямо не связанные с вовлечением нервов – стеноз почечной артерии, легочные кисты и интерстициальная пневмония, неправильное формирование различных отделов желудочно-кишечного тракта, гипертрофия клитора.
Наличие НФ1 у одного или обоих супругов не считается основанием рекомендовать предохранение от беременности.
1. Пятна цвета “кофе с молоком.”
Структура — шванновские клетки, фибробласты, коллаген, тучные клетки.
Озлокачествление — крайне редко.
Лечение – хирургическое или косметологическое (татуировка телесного цвета). Показания к операции — косметический дефект, боли и зуд, быстрое увеличение. Зуд уменьшается при длительном (годы) назначении кетотифена по 2-4 мг в сутки.
2. Интраневральные (плексиформные) нейрофибромы.
Обычно развиваются из чувствительных или симпатических волокон периферического нерва, соответственно часто возможно микрохирургическое удаление с сохранением двигательной функции нерва.
Показания к операции — боли, двигательные нарушения, большие размеры и быстрый рост. Асимптомные опухоли не оперируются.
Озлокачествление — до 5%. При подозрении на малигнизацию производится биопсия. Лечение комбинированное — операция, лучевая и химиотерапия. На конечностях некоторые рекомендуют ампутацию, некоторые – органосохраняющую резекцию.
3. Спинальные нейрофибромы.
Обычно возникают из дорзальных корешков, множественные, встречаются чаще на шейном и поясничном уровнях.
Показания к операции — симптомы сдавления спинного мозга и корешковые, а также большие опухоли, дальнейший рост которых может существенно увеличить операционный риск.
4. Глиомы зрительных нервов.
Наблюдаются у 5 — 10% больных НФ1.
Диагноз устанавливается обычно офтальмологом, требует (особенно у маленьких детей) уточнения с помощью исследования зрительных вызванных потенциалов, верифицируется при КТ или МРТ. На момент постановки диагноза глиомы зрительных нервов при НФ1 у подавляющего большинства больных бывают двусторонними.
Скорость роста этой практически всегда доброкачественной опухоли крайне вариабельна и непредсказуема. Описаны случаи спонтанной регрессии.
Лечение – в большинстве случаев осуществляется динамическое наблюдение или проводится лучевая терапия, без биопсии. Адекватно проведенная лучевая терапия обеспечивает отсутствие прогрессирования опухоли в течение не менее 10 лет у 100% и стабилизацию или улучшение зрения у 80% облученных больных [5]. Однако среднее время реакции на лучевую терапию (т.е. уменьшение размеров опухоли минимум на 50%) составляет около 6 лет.
Соответственно показания к операции возникают при опухолях, формирующих большие интракраниальные узлы со сдавлением диэнцефальных структур, вызывающих внутричерепную гипертензию или значительный экзофтальм.
Тактика при других ассоциированных состояниях:
— Шванномы любого нерва (не двусторонние вестибулокохлеарного) – обычно удаление опухоли.
— Опухоль головного мозга(астроцитомы, менингиомы) — чаще биологически доброкачественные. При наличии клинических проявлений, перитуморозного отека или прогрессировании бессимптомной опухоли – показано ее удаление.
— Односторонний дефект крыши орбиты с пульсирующим экзофтальмом — коррекция по косметическим показаниям.
— Макроцефалия — обычно сочетается с нормотензивной наружной и/или внутренней гидроцефалией, хирургическое лечение нецелесообразно. В редких случаях сочетания с внутричерепной гипертензией производятся операции на ликворной системе.
— Кифосколиоз – встречается примерно у 10% больных, часто требует ранней передней и задней стабилизации позвоночника.
— Передние спинальные менингоцеле – весьма редкая патология, при которой грыжевидные выпячивания твердой мозговой оболочки, часто множественные, внедряются в тела нескольких позвонков, чаще на грудном уровне. Считается показанной стабилизация позвоночника, особенно при появлении спинальной симптоматики.
— Сирингомиелия – при наличии клинических проявлений показано хирургическое лечение.
— Злокачественные опухоли — MPNST, ганглиоглиома, саркома, лейкемия, опухоль Вильма — комплексное лечение.
НФ2 встречается у 1 из 50 000 новорожденных.
Молекулярно-генетические исследования выявили принципиальные отличия в патогенезе НФ1 и НФ2. Это совершенно разные заболевания, требующие дифференцированного клинического подхода.
Ген НФ2 локализуется в 22 хромосоме (22q12) и кодирует синтез другого супрессора опухолевого роста – белка мерлина или шванномина.
НФ2 формально является аутосомно-доминантным генетическим заболеванием.
Возникающие при НФ2 опухоли являются доброкачественными, но более биологически агрессивными, чем при НФ1.
Для установления клинического диагноза НФ2 необходимо выявление:
— или двусторонних неврином VIII нерва (абсолютный диагностический критерий),
— или наличия прямого родственника с НФ2 и
— Либо односторонней невриномы 8 нерва,
— Либо сочетания двух из следующих признаков –
— Нейрофибромы (одной или нескольких).
— Менингиомы (одной или нескольких).
— Глиомы (одной или нескольких).
— Шванномы, включая спинальную (одной или нескольких).
— Ювенильной задней субкапсулярной лентикулярной катаракты или помутнения хрусталика.
Принципиальным отличием опухолей VIII нерва при НФ2 является их гистологическая структура (это только шванномы, тогда как спонтанные и связанные с НФ1 опухоли могут быть и нейрофибромами) и характер роста.
Если не связанные с НФ2 невриномы и нейрофибромы только смещают слуховой нерв, то при НФ2 опухоль в виде виноградных гроздьев часто распространяется между волокнами 8 нерва, что затрудняет сохранение слуха у этих больных. Также при НФ2 затруднено отделение опухоли и от других черепных нервов, в первую очередь — от лицевого.
Показано оперативное лечение этих опухолей.
источник
Нейрофиброматоз – это генетическое заболевание, характерное аномальным ростом клеток-фидеров (шванновских клеток), с образованием мелких и более крупных опухолей.
Опухоли могут быть доброкачественными или злокачественными. Нейрофиброматоз является относительно редким заболеванием, встречается у 1 из 2500-4000 родившегося ребёнка. Это – аутосомно-доминантное наследственное заболевание. Заболевание, обычно, возникает в 2 основных формах (1 и 2 типа, более старые названия – периферический и центральный).
В большинстве случаев в качестве причины развития заболевания играет роль врожденная мутация, но также возможно развитие недуга в результате возникновения новых мутаций. Диагноз может быть точно определён с помощью генетического анализа.
Нейрофиброматоз является наследственным аутосомно-доминантным заболеванием. Это означает, что при определённой комбинации генов болезнь наследуется от родителей. В случае аутосомно-доминантной наследственности оба пола поражаются заболеванием одинаково часто.
Заболевание передаётся генами, расположенными на неполовых хромосомах – аутосомах (17 хромосома наиболее характерна для нейрофиброматоза 1-го типа, 22 – для 2-го).
Другим, менее распространенным, вариантом является появление новых мутаций. Это означает, что заболевание впервые появляется у человека с образованием новых мутаций. Его родители или другие родственники не страдают этим заболеванием, но сам человек недуг впоследствии передает своим потомкам.
Нейрофиброматоз делится на 2 основных типа:
- Тип НФ1, также известный, как болезнь фон Реклингхаузена. Заболевание всего возникает в соотношении 1:3000 человек.
- Тип НФ2 является более редким явлением, постигает 1-го человека из 25000.
Также выделяют еще 4 типа болезни, но они крайне редки и схема их лечения не отличается от терапии заболевания второго типа.
Оба типа образуют отдельные заболевания, которые имеют различные причины и симптомы.
Проявляются оба типа заболевания по-разному и характер клинической картины различен.
Нейрофиброматоз Реклингхаузена проявляется у детей и имеет следующие симптомы:
- Участки кожи характера белого кофе. Светло-коричневые пятна на коже – безболезненные. Пятна в детском возрасте вырастают до 5 мм, в подростковом – увеличиваются в размере до 15 мм.
- Веснушки при этом типе возникают в необычных местах, например, в складках кожи.
- Доброкачественные опухоли кожи – нейрофибромы. Это – доброкачественные опухоли, растущие под кожей. В детстве – маленькие, с увеличением возраста, как правило, становятся больше. Количество нейрофибром варьируется у каждого человека, у некоторых эти опухоли охватывают всё тело. Некоторые из них вызывают постоянный зуд, изменения формы или нарушение функции конечностей.
- Глиома зрительного нерва. Глиома – это доброкачественная опухоль зрительного нерва, вызывающая нарушение зрения, у детей вызывает изменение восприятия цвета.
- Узелки Лиша. Представляют собой коричневые пятна на радужной оболочке глаза.
- Высокое кровяное давление.
- Злокачественные новообразования оболочки периферических нервов. Каждый нерв имеет свою оболочку. Подозрение на злокачественность присутствует, если нейрофиброма вдруг набухает, становится болезненной, появляется слабость, изменения настроения, покалывание в конечностях.
Первые проявления этого типа нарушения обычно происходят после 20-летнего возраста, у детей младшего возраста заболевание, как правило, не вызывает никаких симптомов.
Среди характерной симптоматики отмечают такие нарушения:
- потеря слуха, гудение и покалывание в ушах;
- проблемы с поддержанием равновесия;
- часто присутствует головокружение и рвота;
- рост опухолей в ушной области, которые повреждают слух и равновесие нервов, передающих сигналы в мозг.
У некоторых людей опухоли растут непосредственно в головном мозге, но не всегда проявляются; речь идёт о доброкачественных новообразованиях. Проблема возникает, когда опухоль вырастает до больших размеров и угнетает окружающие ткани мозга. Это может проявиться головной болью, головокружением, рвотой.
Также могут развиваться опухоли спинного мозга, которые могут вызывать:
Доброкачественные опухоли при НФ2 выглядят, как приподнятое кожное покрытие с диаметром около 2 см.
Симптомы нейрофиброматоза других типов:
- заболевание 3 типа характеризуется возникновением ряда нейрофибром кожи, которые могут привести к глиоме зрительного нерва, нейролеммоме и менингиоме;
- 4 тип заболевания является сегментарным, и поражает только одну определённую область кожи;
- нейрофиброматоз 5 типа характеризуется отсутствием нейрофибром и проявляется наличием лишь тёмных пятен;
- 6 тип заболевания характерен появлением (как и в случае нейрофиброматоза 2 типа) после 20-летнего возраста. Возникают нейрофимы; недуг наиболее часто является приобретенным.
При диагностике нейрофиброматоза важно знать, что речь идет о наследственном заболевании с различными проявлениями. На основе этой информации были определены т.н. диагностические критерии, направленные на помощь в «раскрытии» этого заболевания.
Первыми в списке критериев указаны пятна cafe-au-lait («белый кофе»), в отношении которых указано количество 6 штук или больше в размере 5 и более миллиметров. Ещё одна характеристика предполагает наличие 2-х или более нейрофибром, множественные веснушки в складках кожи (в области подмышек и паха) и глиомы зрительного нерва.
Наличие двух и более пятен Лиша и костная дисплазия также играют важную роль в диагностике. И, наконец, не менее важна частота заболевания в семье, особенно, в отношении родителей, братьев и сестёр. Встречаемость вышеуказанных характеристик увеличивается с возрастом пациента.
Чаще всего удаётся диагностировать нейрофиброматоз в детстве, до 4-летнего возраста. Обычно диагноз основывается на типичной клинической картине, но, в случае возникновения сомнений или неопределённости, можно прибегнуть к помощи генетики. При этом для выявления болезни может быть использован анализ ДНК или РНК.
Для этой экспертизы достаточно сбора периферической венозной крови. Когда болезнь возникает у одного из будущих родителей, возможно пренатальное генетическое тестирование, которое часто проводится при амниоцентезе. Кроме того, можно доимплантационно исследовать яйцеклетки или сперму, непосредственно, перед оплодотворением и зачатием ребёнка.
Есть 3 различных типа нейрофиброматоза, которые могут развиться у детей. Каждый из них развивается из-за генетического дефекта, присутствующего в генах или возникающего сразу же после зачатия.
Ген нейрофиброматоза 1 типа присутствует в 17-й хромосоме, и увеличивает выработку белка нейрофибромина. Этот белок помогает контролировать рост клеток в нервной системе. Мутация гена НФ1 приводит к потере белка и клетки растут аномально.
Ген НФ2 присутствует в 22-й хромосоме, и оказывает влияние на продукцию белка Мерлина. Мутация НФ2 приводит к потере белка, вследствие чего доходит к неконтролируемому росту клеток в нервной системе.
Ген SMARCB1 присутствует в 22-й хромосоме и является причиной шванноматоза.
Каждый тип нейрофиброматоза имеет различные признаки и симптомы.
Первый тип заболевания наиболее часто проявляется у ребёнка. Видимые симптомы нейрофиброматоз первого типа у детей включают в себя:
- светло-коричневые кожные высыпания;
На фото характерная картина нейрофиброматоза первого типа у детей
Нейрофиброматоз 2 (НФ2), в основном, поражает уши ребёнка:
- постепенная потеря слуха;
- звон в ушах;
- нарушение равновесия.
В некоторых редких случаях НФ2 может также влиять на спинной мозг и периферийные нервы. Симптомы в этом случае следующие:
- сильная боль;
- онемение или слабость в руках или ногах.
Шванноматоз является редчайшей формой нейрофиброматоза, которая редко встречается у маленьких детей. Этот вариант заболевания, обычно, развивается в старшем возрасте и вызывает возникновение новообразований на позвоночнике, черепно-мозговых или периферических нервах.
В случае присутствия этой формы болезни могут проявляться хронические боли в любой части тела.
Нейрофиброматоз является генетическим заболеванием и, к сожалению, неизлечимым. Образование опухоли происходит на основе врождённых изменений ДНК, и на сегодняшний день нет возможности каким-либо образом повлиять на этот процесс.
Медикаментозное лечение предполагает приём следующих препаратов:
- Кетотифен;
- Фенкарол;
- Тигазон для снижения скорости клеточного деления;
- Лидаза внутримышечно.
В случае возникновения опухоли, которая беспокоит пациента (присутствует давление в области новообразования из-за врастания в здоровую ткань, происходит закрытие желудочно-кишечного тракта или опухоль представляет собой косметически неприятное явление для человека), она может быть удалена хирургическим путём. Обычно хирург пытается удалить всю опухоль. Однако, нет никакой гарантии, что она не появится где-то в другом месте.
Проблематичными являются опухоли в головном мозге, которые могут угнетать его важные области и вызывать разрушительные осложнения зрения, слуха, двигательной системы, являться триггерами паралича или головных болей.
В области головы кроме открытого хирургического вмешательства путём вскрытия черепа и удаления опухоли, также можно выбрать вариант гамма-ножа, действующего на новообразование излучением. Если доброкачественные опухоли перерастают в злокачественные метастазы, может быть рекомендована химиотерапия, лучевая терапия и другие методы, используемые в онкологии.
Лучевая терапия, как правило, исключается по причине возникновения вторичной злокачественности (создание новой опухоли после облучения). Цель лечения заключается в раннем выявлении болезни с диспансеризацией пациента, регулярных проверках и, в случае необходимости, быстром лечении.
Химиотерапия, обычно, является, первым выбором после хирургического удаления. Костная деформация может быть отрегулирована операционным укреплением или косметической корректировкой.
Все вышеперечисленные методы лечения служат только для улучшения качества жизни, облегчения боли или душевных страданий, но не излечивают от этой болезни полностью.
Для лечения нейрофиброматоза народными средствами целители рекомендуют принимать настойку прополиса (100 г прополиса на 500 мл спирта). Настаивается в течение недели в тёмном месте, после чего следует процедить. Принимать ежедневно 3 раза по 30 капель. Настойку хранить в тёмном месте при комнатной температуре.
Жизнь с нейрофиброматозом, в частности первого типа, является очень стрессовой и хлопотной, поскольку это заболевание оказывает разрушительное влияние на внешний вид. Заболевание оказавает негативное психологическое воздействие, даже, если речь идёт о всего лишь небольшом искажении внешнего вида кожи.
Особенно, это касается подростков, которые очень щепетильно относятся к своей внешности, проявления болезни вызывают чувство стыда и приводят к депрессии и тревоге.
Кроме того, осложнения могут быть более серьёзными: опухолевая ткань начинает расти очень быстро, и её клетки распространяются на другие части тела (метастазирование).
Профилактические меры в отношении нейрофиброматоза – это непростой вопрос, включающий в себя несколько вариантов. В случае врожденного заболевания, нет никакого известного стопроцентного способа предотвратить его развитие.
Если случаи нейрофиброматоза имеются в семейном анамнезе, то для пары планирующей ребенка, существует возможность генетического анализа. Необходимо создать семейное дерево и пометить всех уже больных лиц.
Хорошо также знать, о каком типе заболевания идет речь и в рамках пренатальной генетической диагностики обследования будущего ребёнка. Преимплантационное генетическое тестирование в состоянии изучить эмбрион до имплантации в матку.
Если речь идет о приобретенном в течение жизни заболевании, теоретически возможно избегать всего, что может оказать влияние на генетику человека (радиация, химические и токсичные вещества и т.д.), но, к сожалению, без гарантии успеха.
Вышеуказанные возможности профилактики болезни могут показаться чем-то сверхъестественным, но, несмотря на это, они несколько ограничены, и не всегда могут предупредить развитие заболевания на 100%.
источник
ПТГ – полипептид, секретируемый клетками паращитовидных желез. ПТГ синтезируется как 115-аминокислотный прогормон, далее последовательно расщепляется до 84-аминокислотного полипептида. Молекулярные предшественники (прогормоны) ПТГ остаются внутри клеток паращитовидных желез. ПТГ вместе с кальцитонином (его антагонистом) и витамином Д участвует в регуляции обмена кальция и фосфора. Секретируемый ПТГ взаимодействует со специфическим G-протеиновым рецептором II типа, вызывая быстрое увеличение тубулярной реабсорбции кальция в почках и уменьшение реабсорбции фосфора. Длительное повышение уровня в крови ПТГ повышает мобилизацию кальция из костной ткани и увеличивает синтез D-1,25-диоксихолекальциферола в почках, который, в свою очередь, повышает всасывание кальция в кишечнике. ПТГ способствует поступлению кальция и фосфата из костей в кровь, угнетая активность остеобластов; активируя остеоциты и остеокласты, способствует увеличению пула остеокластов. Нормальное изменение уровня гормона характеризуется циркадным ритмом с максимальными значениями в 14–16 ч и снижением до базального уровня в 8 ч утра. Регуляция секреции ПТГ паращитовидными железами осуществляется по принципу обратной связи, регулирующим фактором является содержание кальция в крови. Метаболизируется ПТГ в основном в печени и почках. Время полужизни ПТГ в крови около 4–5 мин.
Деминерализация костной ткани при избытке ПТГ сопровождается увеличением активности ЩФ в крови и повышением выведения оксипролина (специфического компонента коллагена) с мочой из-за резорбции органического матрикса кости под влиянием ПТГ. Значительное увеличение выведения фосфатов с мочой (фосфатурический эффект ПТГ) сопровождается понижением содержания фосфора в крови. Несмотря на некоторое усиление реабсорбции кальция в почечных канальцах под влиянием ПТГ, выделение кальция с мочой из-за нарастающей гиперкальциемии в конечном итоге увеличивается. В очень редких случаях наследственных синдромов резистентности к ПТГ и при почечной недостаточности повышение уровня ПТГ не приводит к увеличению уровня кальция в крови.
- Гиперкальциемия;
- гипокальциемия;
- остеопороз, кистозные изменения костей, остеосклероз тел позвонков;
- мочекаменная болезнь;
- подозрение на множественную эндокринную неоплазию;
- диагностика нейрофиброматоза.
- Гиперпаратиреоз первичный, вторичный;
- псевдогипопаратиреоз;
- эктопическая секреция ПТГ;
- синдром Золлингера-Эллисона;
- флюороз.
- Первичный или вторичный гипопаратироз;
- гипомагниемия;
- гипервитаминоз Д;
- активный остеолиз;
- саркоидоз.
Продолжая использовать наш сайт, вы даете согласие на обработку файлов cookie, пользовательских данных (сведения о местоположении; тип и версия ОС; тип и версия Браузера; тип устройства и разрешение его экрана; источник откуда пришел на сайт пользователь; с какого сайта или по какой рекламе; язык ОС и Браузера; какие страницы открывает и на какие кнопки нажимает пользователь; ip-адрес) в целях функционирования сайта, проведения ретаргетинга и проведения статистических исследований и обзоров. Если вы не хотите, чтобы ваши данные обрабатывались, покиньте сайт.
Copyright ФБУН Центральный НИИ Эпидемиологии Роспотребнадзора, 1998 — 2019
Центральный офис: 111123, Россия, Москва, ул. Новогиреевская, д.3а, метро «Шоссе Энтузиастов», «Перово»
+7 (495) 788-000-1, info@cmd-online.ru
! Продолжая использовать наш сайт, вы даете согласие на обработку файлов cookie, пользовательских данных (сведения о местоположении; тип и версия ОС; тип и версия Браузера; тип устройства и разрешение его экрана; источник откуда пришел на сайт пользователь; с какого сайта или по какой рекламе; язык ОС и Браузера; какие страницы открывает и на какие кнопки нажимает пользователь; ip-адрес) в целях функционирования сайта, проведения ретаргетинга и проведения статистических исследований и обзоров. Если вы не хотите, чтобы ваши данные обрабатывались, покиньте сайт.
источник
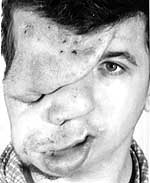
Особенностью заболевания является специфическая последовательность проявления симптомов в зависимости от возраста пациента, что затрудняет клиническую диагностику нейрофиброматоза I типа в раннем детском возрасте. Таким образом, с рождения или первых лет жизни могут существовать лишь некоторые признаки нейрофиброматоза I типа, такие как крупные пигментные пятна, плексиформные нейрофибромы, скелетные дисплазии. Другие симптомы могут проявиться значительно позднее (к 5–15 годам). При этом степень выраженности клинических проявлений, течение и быстрота прогрессирования нейрофиброматоза I типа у разных больных неодинаковы и колеблются в широких пределах. В настоящее время не установлено, чем обусловлены такие различия. Нейрофибромы (дермальные, гиподермальные, плексиформные) представляют собой наиболее выраженное проявление болезни Реклингхаузена, их количество иногда достигает нескольких тысяч; плексиформные нейрофибромы могут быть гигантскими, массой более 10 кг. Эти косметические дефекты, как правило, больше всего беспокоят пациентов, даже имеющих системные заболевания. Кроме того, нейрофибромы, особенно плексиформные, связаны с повышенным риском озлокачествления (в 20% случаев, по нашим данным). При локализации в средостении, в брюшной полости, в глазнице они приводят к нарушению функций прилегающих органов. Например, в сентябре 2000 года в отделении наследственных заболеваний кожи ЦНИКВИ был консультирован больной мальчик 8 лет, прибывший из Брянской области в Москву для оперативного лечения по жизненным показаниям; гигантская плексиформная нейрофиброма располагалась в верхнем средостении, деформировала нижнюю 1/3 шеи и являлась причиной затрудненного дыхания и пароксизмальной тахиаритмии. О развитии нейрофибром известно немногое. Время от времени растет их количество и размеры в ответ на различные стимулы, среди которых ведущее место занимают гормональная перестройка организма: пубертатный возраст, период беременности или после родов, а также перенесенные травмы или тяжелые соматические заболевания. С расширением спектра предлагаемых коммерческих медицинских и косметических услуг значительно увеличилось число обращений больных, указывающих на появление новых опухолей (нейрофибром, неврином, шванном) после ятрогенных вмешательств. Речь идет об удалении опухолей с диагностической или лечебной целью различными методами, в том числе с помощью хирургического иссечения. К ятрогенным осложнениям также приводит назначение физиотерапевтических процедур при лечении различных соматических заболеваний, коррекции скелетных нарушений (всевозможных видов сколиоза, переломов) и нервно-мышечных расстройств (очень часто массаж по различным поводам назначается детям грудного возраста, когда диагностика нейрофиброматоза I типа зачастую невозможна из-за недостаточности клинических проявлений). Но часто заболевание прогрессирует и на фоне кажущегося благополучия. Положение осложняется тем, что у врачей нет возможностей приостановить развитие болезни. Основной задачей научных исследований представляется разработка методов патогенетического лечения нейрофиброматоза I типа, позволяющих сдерживать появление новых и рост уже имеющихся опухолей, а также предотвращать развитие осложнений.
В настоящее время для лечения этого заболевания как за рубежом, так и в России используются методы симптоматической терапии, как, например, хирургическое удаление опухолей, коррекция кифосколиоза или лучевая терапия нейрофибром внутренних органов. Кроме того, ученые на Западе сконцентрировались на возможности этиологического лечения, то есть генной инженерии. Особенно далеко это направление продвинулось со времени открытия мутантного гена и расшифровки его первичного продукта — нейрофибромина в 1990 году; на научные изыскания, связанные с этой проблемой, ежегодно выделяются огромные средства. Первая попытка патогенетического подхода к лечению была сделана V. Riccardi в 1987 г., когда он предложил длительное использование кетотифена (в дозе 2-4 мг в течение 1,5-3 лет) для стабилизации мембран тучных клеток, полагая, что именно дегрануляция этих клеток стимулирует рост опухолей. Однако лечение одним кетотифеном не принесло желаемых результатов: уменьшались субъективные ощущения болезненности и зуда в области нейрофибром, но какого-либо влияния на рост опухолей отмечено не было. Кроме того, при продолжительном приеме препарата наблюдалось снижение количества иммунокомпетентных клеток в периферической крови и ухудшение показателей иммунитета. Существуют различные точки зрения на роль тканевых базофилов в развитии нейрофибром. По мнению некоторых авторов, тучные клетки представляют собой эффекторные клетки противоопухолевого иммунитета. С помощью обычной и электронной микроскопии было показано, что большое количество тканевых базофилов с их активной внеклеточной дегрануляцией наблюдается только на ранней стадии развития нейрофибром. На поздней же стадии, при длительности существования нейрофибром не менее пяти лет, в клеточном матриксе опухолей тучных клеток значительно меньше, дегрануляция их преимущественно внутриклеточная и не сопровождается разрушением клеток. На основании данных многочисленных исследований нами впервые разработана комплексная методика патогенетической терапии с использованием препаратов из разных групп лекарственных средств. Учитывая то, что клеточный состав нейрофибром в основном представлен шванновскими клетками, фибробластами, тучными клетками и лимфоцитами, а межклеточное вещество в активно растущих, особенно плексиформных, — кислыми мукополисахаридами, для лечения нейрофиброматоза I типа мы выбрали следующие препараты. Стабилизатор мембран тучных клеток, кетотифен, мы назначали по 2-4 мг короткими курсами по два месяца. Чтобы избежать осложнений, в первые две недели приема препарата использовался фенкарол по 10-25 мг три раза в день. В качестве антипролиферативного препарата применялись тигазон в дозе не менее 1 мг на килограмм массы тела или аевит до 600 000 МЕ с учетом переносимости. Также курсами применялась лидаза (мукополисахаридаза) в дозе 32-64 Ед в зависимости от возраста внутримышечно, через день, на курс 30 инъекций. Вышеуказанные препараты использовались комплексно в различных сочетаниях или в виде монотерапии в зависимости от формы нейрофиброматоза I типа, жалоб, течения, а также возраста и пола больных. Обязательно лечение проводилось в периоды прогрессирования заболевания, то есть при появлении новых опухолей и/или росте уже имеющихся, как правило сопровождающемся зудом или ощущением болезненности в их проекции, а также с целью предотвращения активизации заболевания во время планируемых операций на опухолях. Повторные курсы лечения с интервалами в два месяца назначались при наличии у больных крупных плексиформных нейрофибром или болезненных неврином. При этом, как правило, курсовое применение тигазона (или аевита) в виде монотерапии чередовалось с сочетанным применением стабилизаторов мембран тучных клеток и инъекций лидазы. Предлагаемое лечение хорошо переносилось больными. В единичных случаях наблюдались незначительное повышение уровней печеночных показателей в повторных биохимических анализах крови при приеме тигазона (у одной больной) и очаговая аллергическая реакция на введение лидазы (у двух пациентов из 60), которая проявлялась воспалением тканей в месте инъекций. В этих случаях препараты отменялись, назначалось симптоматическое лечение. В результате проводимой терапии нам, как правило, удавалось приостановить прогрессирование заболевания; наблюдалось уменьшение (сморщивание) нейрофибром и неврином вплоть до полного исчезновения некоторых опухолей (особенно активно уменьшаются плексиформные нейрофибромы — на ранней стадии своего развития — и невриномы). Полученные результаты удовлетворяют исследователей и позволяют рекомендовать вышеуказанную методику патогенетического лечения нейрофиброматоза I типа для повсеместного применения. Разработанная нами комплексная методика патогенетического лечения впервые дает возможность оказать больным медикаментозную помощь. источник Нейрофиброматоз – болезнь, относящаяся к группе факоматозов. Проявляется в виде пятен телесного цвета на кожных покровах тела. Насчитывают 7 типов нейрофиброматоза (НФ). Рассмотрим причины и признаки каждого типа НФ. Наиболее распространенный тип НФ, который передается наследственным путем. Основной характерной чертой является возникновение опухолей у человека. Заболеваемость в зависимости от пола человека – одинакова, каждый 3,5 тыс. ребенок может подвергаться риску и унаследовать эту болезнь. При НФ-1 в семнадцатой хромосоме картирован ген.
По сравнению с первым типом, процент заболеваемости вторым типом значительно ниже. Примерно каждый 50 тыс. ребенок может заболеть нейрофибрамотозом 2-го типа. При данной разновидности НФ в 22-ой хромосоме картирован ген.
3-тий тип встречается редко.
НФ-3 развивается быстро, но только после 20-30 лет. Довольно редкая форма нейрофиброматоза. Симптоматика схожа с нейрофиброматозом 1-го типа, но есть одно отличие – отсутствуют узелки Лиша. 5-ый тип – сегментарный. При этом типе – поражение одностороннего характера и оно проявляется наличием нейрофибром или пигментных пятен. Иногда эти проявления сочетаются. По своей клинике НФ-5 похож на гемигипертрофию. Данному типу не присущи нейрофибромы. НФ-6 присуще только наличие пигментации, которая проявляется пятнами светло-коричневого цвета. Может образоваться после достижения 20-ти лет. Основные признаки развития:
Наиболее часто болеют нейрофиброматозом первого и второго типа (НФ-1, 2). Для лечения нейрофиброматоза необходимо придерживаться следующих условий:
30 % настойка, приготовленная из корней и листьев болиголова, с корнями аконита, в попеременном применении порошка мухомора (0,2 грамма брать на один прием). Помогает при онкозаболеваниях различного характера. Настой, приготовленный из чистотела. Способ приготовления: 40 грамм травы чистотела поместить в 200 миллилитров кипятка, настоять в течение четверти часа. Пить по 100 миллилитров за четверть часа до еды (не более трех раз в день). Такой настой нельзя хранить больше 48 часов. Внимание! Нужно быть придельно осторожными в применении чистотела для лечения. Ведь, кроме своих лечебных способностей, он известен как ядовитое растение (поэтому нужно строго придерживаться доз). Настои или отвары из корня и стебля лопуха, а, также, лопуха паутинистого. Способ применения: внутрь, трижды в день по 100 миллилитров такого настоя (отвара).
Все эти продукты вызывают интоксикацию организма, нарушается питание подкожных клеток, разрушают клетки кожи. источник | ||||||||||
 Нейрофиброматоз I типа (классический, периферический, собственно болезнь Реклингхаузена) — это тяжелое системное наследственное заболевание с преимущественным поражением кожи и нервной системы, одно из наиболее распространенных моногенных заболеваний человека, встречающееся с частотой не реже 1:3000 — 1:4000 населения. Наследуется аутосомно-доминантно, с высокой пенетрантностью и вариабельной экспрессивностью. Заболевание обусловлено мутацией гена «нф1» в 17q-хромосоме. Мужчины и женщины поражаются одинаково часто. Примерно половина случаев — следствие новых мутаций.
Нейрофиброматоз I типа (классический, периферический, собственно болезнь Реклингхаузена) — это тяжелое системное наследственное заболевание с преимущественным поражением кожи и нервной системы, одно из наиболее распространенных моногенных заболеваний человека, встречающееся с частотой не реже 1:3000 — 1:4000 населения. Наследуется аутосомно-доминантно, с высокой пенетрантностью и вариабельной экспрессивностью. Заболевание обусловлено мутацией гена «нф1» в 17q-хромосоме. Мужчины и женщины поражаются одинаково часто. Примерно половина случаев — следствие новых мутаций.